

近日,動物醫學院王林教授團隊在《Journal of Advanced Research》發表了題為“Cadmium targeting transcription factor EB to inhibit autophagy-lysosome function contributes to acute kidney injury”的研究論文。動物醫學院已畢業碩士生董鵬飛為該論文第一作者,王林教授為該論文的通訊作者。
鎘(Cd)是生態環境中一種常見的重金屬污染物,主要通過食物鏈的富集作用對人類和動物健康造成嚴重危害。Cd也是一種蓄積性毒物,對機體的損傷呈多系統性和多器官性。腎臟是鎘最重要的蓄積部位和靶器官,職業性鎘暴露可引起急性腎損傷。目前AKI的發病機制復雜,其中自噬-溶酶體功能障礙在AKI發病過程中發揮重要作用。轉錄因子EB(TFEB)已被證明是溶酶體生物發生和自噬相關基因轉錄的關鍵調控因子。然而,TFEB在Cd誘導AKI中的潛在機制尚不清楚。因此,闡明TFEB在Cd所誘導自噬-溶酶體功能障礙中的作用對于緩解AKI具有重要意義。如圖1結果所示,Cd誘導的腎小管上皮細胞損傷和自噬-溶酶體功能障礙可通過TFEB的藥理激活劑或基因過表達得到緩解,而沉默TFEB基因則加劇其損傷。本研究結果強調了Cd通過抑制TFEB功能導致自噬-溶酶體功能障礙而促進AKI的病理學進程。
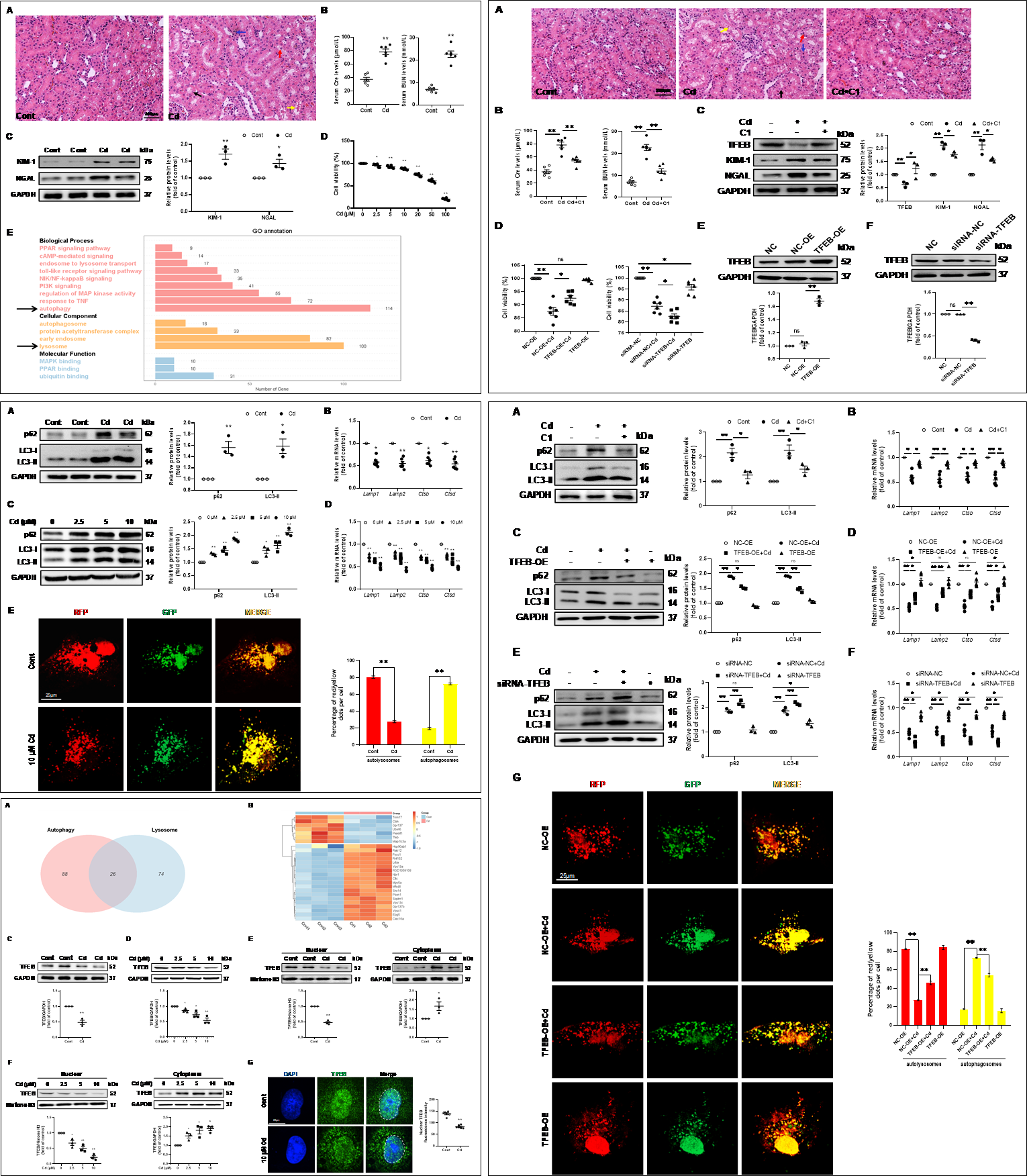
圖1 TFEB依賴性自噬-溶酶體功能障礙促進Cd誘導的急性腎損傷
從機制上講,Cd激活雷帕霉素復合體1(mTORC1)增強TFEB磷酸化,從而抑制TFEB核易位;Cd也激活染色體區域維持1(CRM1)促進TFEB核輸出。同時,Cd激活組蛋白乙酰轉移酶GCN5增強核TFEB乙酰化,導致TFEB轉錄活性降低。研究結果表明,Cd對TFEB活性的抑制依賴于mTORC1介導的TEFB磷酸化、CRM1介導的TFEB核輸出和GCN5介導的TFEB乙酰化的聯合作用,但TFEB磷酸化、核輸出和乙酰化分別通過抑制TFEB活性促進自噬-溶酶體功能障礙,共同參與了Cd誘導的AKI。這些研究結果解析了Cd誘導腎損傷的潛在分子機制(圖2),闡明了TFEB是Cd誘導AKI的一個潛在治療靶點。
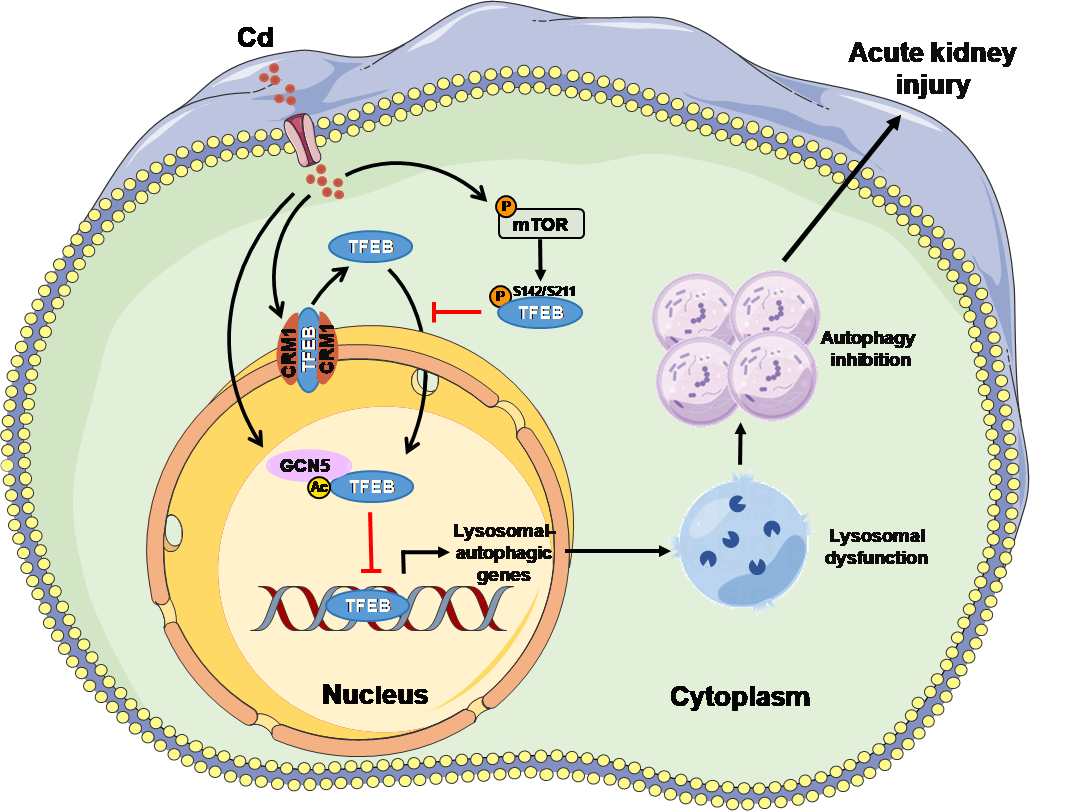
圖2 Cd靶向TFEB抑制自噬-溶酶體功能導致AKI的示意圖
本研究得到了國家自然科學基金面上項目和山東省高等學校青創科技計劃創新團隊項目資助。
全文鏈接:https://doi: 10.1016/j.jare.2024.07.013.
編 輯:萬 千
審 核:賈 波





